Tiêu chuẩn quốc gia TCVN 1676-1:2007 (ISO 5418-1 : 2006) về Quặng sắt - Xác định đồng - Phần 1: phương pháp đo màu 2,2’-biquinolyl
TIÊU CHUẨN QUỐC GIA
TCVN 1676-1 : 2007
ISO 5418-1 : 2006
QUẶNG SẮT - XÁC ĐỊNH ĐỒNG - PHẦN 1: PHƯƠNG PHÁP ĐO MÀU 2,2’-BIQUINOLYL
Iron ores - Determination of copper – Part 1: 2,2’-Biquinolyl spectrophotometric method
Lời nói đầu
TCVN 1676-1 : 2007 và TCVN 1676-2 : 2007 thay thế TCVN 1676 : 1986
TCVN 1676-1 : 2007 hoàn toàn tương đương với ISO 5418-1 : 2006.
TCVN 1676-1 : 2007 do Tiểu ban Kỹ thuật Tiêu chuẩn TCVN/TC102/SC2 Quặng sắt – Phân tích hoá học biên soạn, Tổng cục Tiêu chuẩn Đo lường Chất lượng đề nghị, Bộ Khoa học và Công nghệ công bố TCVN 1676 (ISO 5418) với tên chung là: Quặng sắt – Xác định đồng gồm hai phần:
– Phần 1: Phương pháp đo màu 2,2’-biquinolyl;
– Phần 2: Phương pháp quang phổ hấp thụ nguyên tử ngọn lửa.
QUẶNG SẮT - XÁC ĐỊNH ĐỒNG – PHẦN 1: PHƯƠNG PHÁP ĐO MÀU 2,2’-BIQUINOLY
Iron ores – Determination of copper – Part 1: 2,2’-Biquinoly spectrophotometric method
Cảnh báo Tiêu chuẩn này có thể liên quan đến các vật liệu, thao tác và thiết bị nguy hại. Tiêu chuẩn này không đề cập đến những vấn đề về an toàn liên quan đến việc sử dụng chúng. Trách nhiệm của người sử dụng tiêu chuẩn này là phải thiết lập các quy tắc phù hợp về sức khoẻ, an toàn và xác định các giới hạn cho phép trước khi sử dụng.
1. Phạm vi áp dụng
Tiêu chuẩn này quy định phương pháp đo màu 2,2’- biquinolyl để xác định đồng trong quặng sắt. Phương pháp này có thể áp dụng cho dải hàm lượng đồng từ 0,005 % đến 0,77 % khối lượng trong quặng sắt nguyên khai, tinh quặng và sắt kết khối, kể cả các sản phẩm thiêu kết.
2. Tài liệu viện dẫn
Các tài liệu viện dẫn sau là cần thiết cho việc áp dụng tiêu chuẩn này. Đối với các tài liệu viện dẫn ghi năm ban hành thì áp dụng bản được nêu. Đối với các tài liệu viện dẫn không ghi năm ban hành thì áp dụng phiên bản mới nhất, bao gồm cả các sửa đổi (nếu có).
TCVN 1664 : 2007 (ISO 7764 : 2006) Quặng sắt – Chuẩn bị mẫu thử đã sấy sơ bộ để phân tích hoá học.
TCVN 4851 (ISO 3696) Nước dùng để phân tích trong phòng thí nghiệm – Yêu cầu kỹ thuật và phương pháp thử.
TCVN 7151 (ISO 648) Dụng cụ thí nghiệm bằng thủy tinh – Pipet một mức. TCVN 7153 (ISO 1042) Dụng cụ thí nghiệm bằng thủy tinh – Bình định mức.
ISO 3082 Iron ores – Sampling and sample preparation procedures (Quặng sắt – Quy trình lấy mẫu và chuẩn bị mẫu).
3. Nguyên tắc
Phân hủy phần mẫu thử bằng axit clohydric, axit nitric và axit pecloric.
Khử nước của silica và pha loãng dung dịch rồi lọc. Nung phần cặn, xử lý bằng axit fluohydric và sulfuric, rồi làm nóng chảy với natri cacbonat. Chất chảy đã nguội được hoà tan trong dung dịch lọc.
Đồng (II) được khử bằng axit ascobic. Thêm 2,2’-biquinolyl với sự có mặt của N,N-dimetyl formamid để tạo thành phức đồng (I) mầu tím đỏ.
Mức độ hấp thụ của phức mầu được xác định bằng phương pháp đo màu tại bước sóng khoảng 545 nm.
4. Thuốc thử
Trong quá trình phân tích, chỉ sử dụng thuốc thử tinh khiết phân tích và nước phù hợp với loại 3 của TCVN 4851 (ISO 3696).
Dụng cụ chưng cất không được chứa đồng và nước đã khử ion, không được tiếp xúc với ống hoặc vòi đồng.
4.1 Natri cacbonat (Na2CO3), bột khan.
4.2 Sắt (III) ôxit, độ tinh khiết tối thiểu: 99,9 % khối lượng, có hàm lượng đồng nhỏ hơn 0,0002 % khối lượng.
4.3 Axit clohydric, r 1,16 g/ml đến 1,19 g/ml.
4.4 Axit clohydric, r 1,16 g/ml đến 1,19 g/ml, pha loãng 1 + 2.
4.5 Axit clohydric, r 1,16 g/ml đến 1,19 g/ml, pha loãng 1 + 10.
4.6 Axit nitric, r 1,4 g/ml.
4.7 Axit nitric, r 1,4 g/ml, pha loãng 1 + 1.
4.8 Axit pecloric, r 1,54 g/ml, 60 % khối lượng hoặc r 1,67 g/ml, 70% khối lượng.
4.9 Axit sulfuric, r 1,84 g/ml, pha loãng 1 + 1.
4.10 Axit fluohydric, r 1,13 g/ml, 40 % khối lượng hoặc r 1,185 g/ml, 48% khối lượng.
4.11 Axit ascobic (C6H806) dung dịch, 200 g/l. Chuẩn bị dung dịch này trước lúc sử dụng.
4.12 N,N-dimetylformamid [HCON(CH3)2].
Cảnh báo Phải cẩn thận tránh hít phải khói độc.
4.13 2,2’-biquinolyl (C18H12N2) dung dịch.
Hoà tan 0,15 g 2,2’-biquinolyl trong 250 ml N,N-dimetylformamid. Bảo quản dung dịch tránh ánh sáng và bảo quản trong chai màu nâu.
4.14 Dung dịch đồng tiêu chuẩn
4.14.1 Dung dịch tiêu chuẩn A, 1 000 mg Cu/ml.
Hoà tan 0,500 g kim loại đồng (độ tinh khiết tối thiểu 99,9 % khối lượng) trong 20 ml axit nitric loãng (4.7) trong cốc thành cao dung tích 250 ml. Sau khi loại trừ khói nitơ oxit bằng cách đun sôi, làm nguội, chuyển sang bình định mức 500 ml, pha loãng bằng nước đến thể tích rồi lắc đều.
4.14.2. Dung dịch tiêu chuẩn B, 50 mg Cu/ml.
Chuyển 25,0 ml dung dịch tiêu chuẩn A (4.14.1) sang bình định mức 500 ml pha loãng bằng nước đến thể tích.
5. Thiết bị, dụng cụ
Thiết bị phòng thí nghiệm thông thường, bao gồm pipet một vạch mức, bình định mức phù hợp với các quy định của TCVN 7151 (ISO 648) và TCVN 7153 (ISO 1042) (ngoại trừ các chỉ dẫn khác) và các thiết bị, dụng cụ sau.
5.1. Chén bạch kim, dung tích từ 25 ml đến 30 ml.
5.2. Lò múp, phù hợp để nung ở 1 000 oC
5.3. Máy đo màu, phù hợp để đo độ hấp thụ ở bước sóng khoảng 545 nm.
6. Lấy mẫu và mẫu thử
6.1. Quy định chung
Để phân tích, sử dụng mẫu phòng thí nghiệm có cỡ hạt nhỏ hơn 100 mm được lấy và chuẩn bị theo ISO 3082. Trong trường hợp quặng có hàm lượng nước liên kết đáng kể hoặc các hợp chất có thể bị ôxy hoá, sử dụng cỡ hạt nhỏ hơn 160 mm.
Chú thích Hướng dẫn về hàm lượng nước liên kết đáng kể và các hợp chất có thể bị ôxy hoá theo TCVN 1664 : 2007 (ISO 7764 : 2006).
6.2. Chuẩn bị mẫu thử đã sấy sơ bộ
Trộn đều mẫu phòng thí nghiệm và tiến hành lấy các mẫu đơn, từ đó lấy ra các mẫu thử sao cho đảm bảo tính đại diện cho toàn bộ mẫu trong thùng. Sấy mẫu thử ở 105 oC ± 2 oC theo TCVN 1664 : 2007 (ISO 7764 : 2006). (Đây là mẫu thử đã sấy sơ bộ).
7. Cách tiến hành
7.1. Số phép xác định
Tiến hành phân tích độc lập ít nhất hai phép xác định trên một mẫu thử đã sấy sơ bộ, theo Phụ lục A.
Chú thích Khái niệm “độc lập” có nghĩa là kết quả thứ hai và bất kỳ kết quả ngoại suy nào không bị ảnh hưởng bởi các kết quả trước. Đối với phương pháp phân tích cụ thể này, điều kiện này hàm ý là việc tái diễn quy trình được thực hiện do cùng người thao tác tại thời điểm khác hoặc do một người thao tác khác, kể cả việc hiệu chuẩn lại thích hợp trong mỗi trường hợp.
7.2 Phần mẫu thử
Lấy một số mẫu đơn, cân khoảng 0,5 g hoặc 1 g mẫu thử chính xác đến 0,000 2 g (xem Bảng 1) thu được theo 6.2.
Thao tác lấy và cân phần mẫu thử phải nhanh để tránh hấp thụ ẩm lại.
Bảng 1 – Hướng dẫn phép đo dung dịch thử
|
Hàm lượng đồng trong mẫu thử, % khối lượng |
Khối lượng phần mẫu thử, g |
Bình định mức, ml |
Cuvet, cm |
|
0,004 đến 0,05 0,05 đến 0,4 0,4 đến 0,8 |
1,0 0,5 0,5 |
50 100 100 |
5 2 1 |
7.3 Phép thử trắng và phép thử kiểm tra
Trong mỗi loạt phép thử, tiến hành song song một phép thử trắng và một phép thử chất chuẩn được chứng nhận cùng loại với mẫu quặng trong cùng một điều kiện. Mẫu thử đã sấy sơ bộ của chất chuẩn được chứng nhận phải chuẩn bị như quy định tại 6.2.
Chất chuẩn được chứng nhận phải cùng loại với mẫu phân tích và tính chất của hai mẫu phải gần giống nhau để đảm bảo, trong cả hai trường hợp, không cần phải có sự thay đổi đáng kể trong quy trình phân tích.
Khi thực hiện phân tích vài mẫu cùng lúc, có thể sử dụng giá trị phép thử trắng cho một lần thử, với điều kiện sử dụng cùng quy trình và sử dụng cùng chai thuốc thử.
Khi thực hiện phân tích cùng lúc vài mẫu của cùng loại quặng, có thể dùng chung kết quả phân tích của chất chuẩn được chứng nhận.
7.4. Phép xác định
7.4.1. Phân hủy phần mẫu thử
Chuyển phần mẫu thử (7.2) vào cốc thành cao dung tích 250 ml, tẩm ướt bằng 5 ml nước. Thêm 20 ml axit clohydric (4.3), đậy cốc bằng nắp kính đồng hồ, đun nóng nhẹ dung dịch nhưng không sôi cho đến khi phân hủy hoàn toàn mẫu thử. Thêm 5 ml axit nitric (4.6) tiếp theo là 10 ml axit percloric (4.8) và 0,2 ml axit sulfuric (4.9), đậy cốc bằng nắp kính đồng hồ, đun nóng đến khi khói axit percloric bay ra. Tiếp tục đun nóng thêm 3 phút đến 5 phút.
Để cốc nguội rồi thêm 20 ml axit clohydric (4.4). Đun sôi trong 1 phút để loại bỏ clo, pha loãng bằng 10 ml nước.
Lọc dung dịch qua giấy lọc trung bình, gộp nước lọc vào trong cốc 300 ml. Tia rửa giấy lọc bằng axit clohydric (4.5), sử dụng thể tích càng nhỏ càng tốt, cho đến khi màu vàng của sắt (III) biến mất. Cuối cùng, tia rửa bằng nước nóng tới khi nước rửa không còn axit. Bảo quản nước lọc và nước rửa như là dung dịch chính. Chuyển giấy lọc chứa cặn vào chén bạch kim (5.1).
7.4.2 Xử lý cặn
Sấy và tro hoá giấy lọc ở nhiệt độ thấp, và nung cặn ở nhiệt độ khoảng 800 oC trong lò múp (5.2).
Để chén nguội, tẩm ướt cặn bằng một vài giọt nước, rồi thêm 5 giọt axit sulfuric (4.9) và 5 ml axit flohydric (4.10).
Đun nhẹ trong tủ hút để làm bay hơi silic ở dạng tetraflorit và cô axit sulfuric đến khô. Cuối cùng đun chén ở nhiệt độ cao trong vài giây để đảm bảo tách hoàn toàn axit sulfuric. Để chén nguội và thêm 1 g natri cacbonat (4.1). Nung nhẹ trong vài phút sau đó ở nhiệt độ từ 900 oC đến 1 000 oC cho đến khi cặn phân hủy hoàn toàn.
Chú thích Nếu nhiều cặn, có thể thêm natri cacbonat. Như vậy, số lượng natri cacbonat lấy theo 7.5 phải tăng lên.
Để chén nguội rồi chuyển vào cốc chứa dung dịch chính ở 7.4.1, đun nhẹ để hoà tan khối chảy. Lấy chén ra và rửa bằng nước. Cô dung dịch nếu cần và làm nguội đến nhiệt độ phòng. Chuyển sang bình định mức 50 ml hoặc 100 ml như chỉ dẫn trong Bảng 1, pha loãng bằng nước đến thể tích và lắc đều. (Đây là dung dịch thử).
7.4.3 Xử lý dung dịch thử
Chuyển 10,0 ml dung dịch ở 7.4.2 vào hai bình định mức 50 ml. Thêm các thuốc thử sau, lắc đều sau mỗi lần thêm:
- 5 ml dung dịch axit ascobic (4.11) và 25 ml dung dịch 2,2’-biquinolyl (4.13), đối với dung dịch thử;
- 5 ml dung dịch axit ascobic (4.11) và 25 ml dung dịch N,N-dimetylformamid (4.12) đối với dung dịch chuẩn.
Tương tự, chuyển 10 ml dung dịch thử trắng vào hai bình định mức 50 ml. Vừa thêm vừa lắc các thuốc thử sau:
- 5 ml dung dịch axit ascobic (4.11) và 25 ml dung dịch 2,2’-biquinolyl (4.13) đối với dung dịch thử trắng;
- 5 ml dung dịch axit ascobic (4.11) và 25 ml dung dịch N,N-dimetyl formamid (4.12) đối với dung dịch chuẩn trắng.
Pha loãng từng dung dịch đến thể tích bằng nước, lắc đều và đặt các bình trong bể nước ở khoảng 20 oC trong 5 phút. Điều chỉnh vạch mức, nếu cần, lắc đều, để yên trong 10 phút rồi đo.
7.4.4 Phép đo màu
Sử dụng bước sóng phù hợp (xem Bảng 1), đo sự hấp thụ của dung dịch thử theo dung dịch đối chứng. Bước sóng hấp thụ lớn nhất khoảng 545 nm.
Tương tự, đo sự hấp thụ của dung dịch thử trắng theo dung dịch trắng đối chứng trong cùng điều kiện.
Hiệu chỉnh giá trị hấp thụ của dung dịch thử với giá trị hấp thụ thu được đối với dung dịch thử trắng.
7.5 Chuẩn bị đường chuẩn
Cân 0,5 g hoặc 1,0 g sắt (III) oxit (4.2) theo Bảng 2, chuyển vào cốc thành cao 250 ml, và hoà tan trong 20 ml axit clohydric (4.3).
Thêm lượng dung dịch chuẩn đồng A (4.14.1) hoặc B (4.14.2) theo Bảng 2.
Thêm 5 ml axit nitric (4.6), 0,2 ml axit sulfuric (4.9) và 10 ml axit percloric (4.8) vào mỗi bình. Đun nóng đến khi xuất hiện khói axit percloric rồi đun tiếp trong 3 phút đến 5 phút.
Để nguội và thêm 20 ml axit clohydric (4.4). Cẩn thận thêm 1 g natri cacbonat (4.1), đun sôi trong 1 phút để tách clo và cacbon dioxit, rồi làm nguội đến nhiệt độ phòng.
Chuyển dung dịch số 1 đến 4 vào bốn bình định mức 50 ml, và dung dịch số 5 đến 11 vào bảy bình định 100 ml. Pha loãng bằng nước đến thể tích rồi lắc đều.
Tiếp tục như chỉ dẫn trong 7.4.3 và 7.4.4. Dựng đồ thị tương quan giữa khối lượng đồng và độ hấp thụ.
Chú thích Dung dịch hiệu chuẩn số 1 (không thêm đồng) được sử dụng như dung dịch trắng đối với hàm lượng đồng từ 0,004 % đến 0,05 % khối lượng, còn dung dịch hiệu chuẩn số 5 (không thêm đồng) được sử dụng đối với hàm lượng đồng từ 0,05 % đến 0,8 % khối lượng.
Hàm lượng đồng tính theo khối lượng liên quan đến khối lượng phần mẫu thử 1 g hoặc 0,5 g trong các điều kiện nêu trong Bảng 1.
Bảng 2 – Dung dịch hiệu chuẩn
|
Dung dịch số |
Khối lượng sắt (III) ôxit, g |
Thể tích dung dịch chuẩn đồng, ml |
Đồng mg |
Đồng % |
|
|
A |
B |
||||
|
1 2 3 4 5 6 7 8 9 10 11 |
1,0 1,0 1,0 1,0 0,5 0,5 0,5 0,5 0,5 0,5 0,5 |
0 2,0 3,0 4,0 |
0 1,0 5,0 10,0 0 5,0 10,0 20,0 |
0 0,05 0,25 0,50 0 0,25 0,50 1,00 2,00 3,00 4,00 |
0 0,005 0,025 0,050 0 0,05 0,10 0,20 0,40 0,60 0,80 |
8. Biểu thị kết quả
8.1. Tính hàm lượng đồng
Hàm lượng đồng, wCu, tính bằng phần trăm theo công thức
wCu = ![]()
trong đó
mo là khối lượng của phần mẫu thử, tính bằng gam;
m1 là khối lượng đồng chứa trong dung dịch lấy trong 7.4.3 và xác định từ đồ thị chuẩn, tính bằng microgam;
f là hệ số pha loãng (f = 0,5 nếu sử dụng phần mẫu thử là 1 g, các trường hợp còn lại f = 1);
V là thể tích dung dịch lấy trong 7.4.5, tính bằng mililit.
8.2. Xử lý chung các kết quả
8.2.1. Độ lặp lại và sai số cho phép
Độ chụm của phương pháp phân tích biểu thị bằng các phương trình hồi quy sau:
sd = 0,009 5X 0,5779 (2)
sL = 0,013 3X 0,5201 (3)
Rd = 0,026 8X 0,5779 (4)
P = 0,042 5X 0,5305 (5)
trong đó
Rd là giới hạn kết quả song song độc lập;
P là sai số cho phép giữa các phòng thí nghiệm;
sd là độ lệch chuẩn của kết quả song song độc lập;
sL là độ lệch chuẩn giữa các phòng thí nghiệm;
X là hàm lượng đồng trong mẫu thử đã sấy sơ bộ, biểu thị bằng phần trăm khối lượng được tính như sau:
- với công thức (2) và (3) trong phòng thí nghiệm: trung bình của kết quả song song;
- với công thức (4) và (5) giữa các phòng thí nghiệm: trung bình kết quả cuối cùng
(8.2.3) của hai phòng thí nghiệm.
8.2.2. Xác định kết quả phân tích
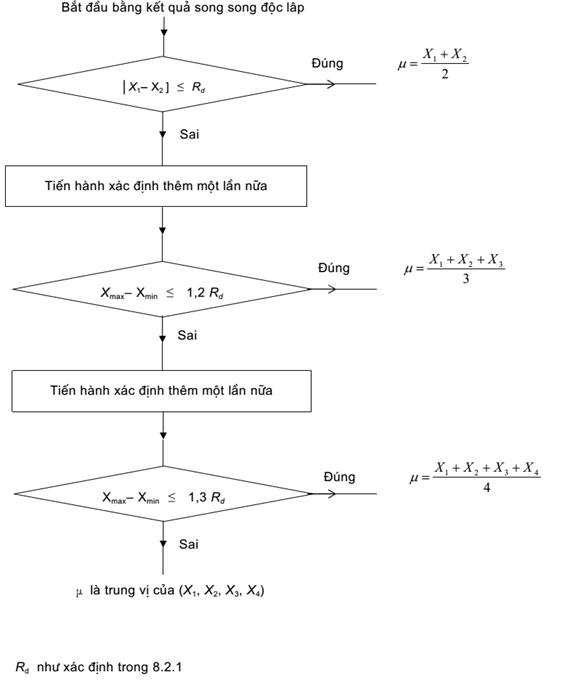
Sau khi tính được các kết quả song song độc lập theo phương trình (1), so sánh với kết quả giới hạn song song độc lập (Rd), sử dụng quy trình nêu trong Phụ lục A
8.2.3. Độ chụm giữa các phòng thí nghiệm
Độ chụm giữa các phòng thí nghiệm được sử dụng để xác định sự phù hợp của kết quả cuối cùng giữa hai phòng thí nghiệm. Giả thiết rằng hai phòng thí nghiệm tiến hành cùng phương pháp đã mô tả trong 8.2.2.
Tính đại lượng sau
m12 = ![]()
trong đó
m1 là kết quả cuối cùng của phòng thí nghiệm 1;
m2 là kết quả cuối cùng của phòng thí nghiệm 2;
m12 là kết quả trung bình của hai phòng thí nghiệm.
Nếu |m1 - m2| ≤ P, kết quả cuối cùng được chấp nhận.
8.2.4. Kiểm tra độ đúng
Độ đúng của phương pháp phân tích phải được kiểm tra bằng cách sử dụng chất chuẩn được chứng nhận (CRM) hoặc chất chuẩn (RM). Quy trình giống như đã mô tả ở trên. Sau khi đáp ứng độ chính xác, kết quả cuối cùng được so sánh với giá trị chuẩn hoặc giá trị chuẩn chứng nhận Ac có hai khả năng:
a) |mc - Ac| ≤ C trong trường hợp này, chênh lệch giữa kết quả phân tích và giá trị chứng nhận/chuẩn không có ý nghĩa về thống kê;
b) |mc - Ac| > C trong trường hợp này, chênh lệch giữa kết quả phân tích và giá trị chứng nhận/chuẩn có ý nghĩa về thống kê;
trong đó
mc là kết quả cuối cùng của CRM/RM;
Ac là giá trị chuẩn/chứng nhận của CRM/RM;
C là giá trị phụ thuộc vào loại mẫu chuẩn CRM/RM được sử dụng.
Các chất chuẩn được chứng nhận sử dụng cho mục đích này phải được chuẩn bị và chứng nhận theo ISO Guide 35 Chứng nhận chất chuẩn – Nguyên tắc chung và nguyên tắc thống kê.
Đối với mẫu CRM/RM do chương trình thử nghiệm liên phòng chứng nhận.
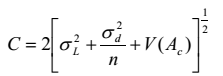
trong đó
V(Ac) là phương sai của giá trị chứng nhận/chuẩn Ac (= 0 với CRM/RM do một phòng thí nghiệm chứng nhận);
n là số phép thử lặp lại đã tiến hành trên CRM/RM.
Tránh sử dụng CRM do một phòng thí nghiệm chứng nhận, trừ khi biết được giá trị chứng nhận không có độ chệch.
8.2.5. Tính kết quả cuối cùng
Kết quả cuối cùng là trung bình số học của các giá trị phân tích được chấp nhận cho mẫu thử, hoặc như được xác định theo quy định trong Phụ lục A, tính đến năm số thập phân và làm tròn đến số thập phân thứ ba như sau:
a) Khi số thập phân thứ tự nhỏ hơn 5 thì bỏ đi và giữ nguyên số thập phân thứ ba;
b) Khi số thập phân thứ tự bằng 5 và số thập phân thứ năm khác 0, hoặc số thập phân thứ tự lớn hơn 5 thì tăng số thập phân thứ ba lên một đơn vị;
c) Khi số thập phân thứ tự bằng 5 và số thập phân thứ năm bằng 0 thì bỏ số 5 và giữ nguyên số thập phân thứ ba khi nó là 0, 2, 4, 6 hoặc 8 và tăng lên một đơn vị khi nó là 1, 3, 5, 7 hoặc 9.
8.3. Tính hàm lượng đồng oxit
Hàm lượng đồng oxit, biểu thị bằng phần trăm được xác định theo phương trình sau:
wCuO = 1,251 8 wCu
9. Báo cáo thử nghiệm
Báo cáo thử nghiệm gồm các thông tin sau:
a) tên và địa chỉ phòng thử nghiệm;
b) ngày tháng báo cáo kết quả;
c) viện dẫn tiêu chuẩn này;
d) các chi tiết cần thiết để nhận biết mẫu;
e) kết quả phân tích;
f) số tham chiếu của phiếu kết quả;
g) bất kỳ các đặc điểm đã ghi nhận trong quá trình xác định, các thao tác không quy định trong tiêu chuẩn này có thể ảnh hưởng đến kết quả của mẫu thử hoặc chất chuẩn được chứng nhận.
Phụ lục A
(quy định)
Lưu đồ quy trình chấp nhận giá trị phân tích đối với mẫu thử
Phụ lục B
(tham khảo)
Nguồn gốc của các phương trình độ lặp lại và sai số cho phép
Các phương trình trong 8.2.1 có nguồn gốc từ những kết quả các loạt phân tích quốc tế tiến hành trong năm 1972 và 1973 của năm mẫu quặng, do 37 phòng thí nghiệm của tám quốc gia thực hiện.
Xử lý đồ thị các số liệu độ chính xác nêu trong Phụ lục C. Các mẫu thử đã sử dụng được liệt kê trong Bảng B.1.
Bảng B.1 – Khối lượng đồng trong mẫu thử
|
Mẫu |
Hàm lượng đồng, % khối lượng |
|
Tinh quặng Kiruna R Bã nung Tro pyrit Nga Forsbo |
0,001 2 0,011 5 0,067 7 0,376 0,772 |
Chú thích 1 Báo cáo thử nghiệm quốc tế và phân tích thống kê các kết quả (tài liệu ISO/TC102/SC2 N313E, tháng 10 1973) được lưu tại Ban thư ký ISO/TC 102/SC2.
Chú thích 2 Đánh giá thống kê được trình bày theo nguyên tắc của TCVN 6910-2 (ISO 5725-2) Độ chính xác (độ đúng và độ chụm) của phương pháp đo và kết quả đo – Phần 2: Phương pháp cơ bản xác định độ lặp lại và độ tái lập của phương pháp đo tiêu chuẩn.
Phụ lục C
(tham khảo)
Dữ liệu độ chụm thu được do các thử nghiệm phân tích quốc tế
Chú thích Hình C.1 biểu thị bằng đồ thị các phương trình trong 8.2.1
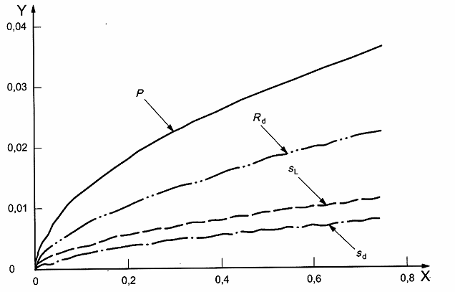
Chú giải
X Hàm lượng đồng, %
Y Độ chụm, %
Hình C.1 – Tương quan bình phương tối thiểu của độ chụm so với hàm lượng đồng