Tiêu chuẩn Việt Nam TCVN 6258:1997 (ASTM D 2795) về Than và cốc - Phương pháp thử chuẩn để phân tích tro do Bộ Khoa học Công nghệ và Môi trường ban hành
TIÊU CHUẨN VIỆT NAM
TCVN 6258 : 1997
ASTM D 2795
THAN VÀ CỐC - PHƯƠNG PHÁP THỬ CHUẨN
ĐỂ PHÂN TÍCH TRO
Standard test methods for analysis of coal and coke ash
Lời nói đầu
TCVN 6258 : 1997 hoàn toàn tương đương với ASTM D 2795.
TCVN 6258 : 1997 do Ban kỹ thuật TCVN/TC 27 “Nhiên liệu khoáng rắn” biên soạn, Tổng cục Tiêu chuẩn – Đo lường – Chất lượng đề nghị, Bộ Khoa học – Công nghệ và Môi trường ban hành.
THAN VÀ CỐC - PHƯƠNG PHÁP THỬ CHUẨN
ĐỂ PHÂN TÍCH TRO
Standard test methods for analysis of coal and coke ash
1.Phạm vi áp dụng
1.1 Tiêu chuẩn này qui định các phương pháp phân tích nhanh và không tốn kém để phân tích những thành phần chính thường được xác định trong tro của than và cốc.
1.2 Các phương pháp thực hiện theo trình tự sau:
Các điều
Silic dioxit (SiO2) từ 10 đến 12
Nhôm oxit (Al2O3) từ 13 đến 15
Sắt oxit (Fe2O3) từ 16 đến 18
Titan dioxit (TiO2) từ 19 đến 22
Photpho pentoxit (P2O5) từ 23 đến 25
Canxi oxit (CaO) và magiê oxit (MgO) từ 26 đến 29
Natri oxit (Na2O) và kali oxit (K2O) từ 30 đến 33
Chú thích 1 - ASTM D1757 được dùng để xác định lưu huỳnh.
1.3 Các trị số được biểu thị ở đơn vị SI được coi là tiêu chuẩn. Các trị số đặt trong ngoặc đơn là để thông tin.
1.4 Tiêu chuẩn này không chủ định đề cập đến tất cả những vấn đề an toàn. Nếu có vấn đề liên quan đến an toàn, người áp dụng tiêu chuẩn này phải xác định sự an toàn phù hợp. Thực tiễn sức khỏe và khả năng áp dụng các giới hạn thường được xác định trước khi sử dụng.
2. Tiêu chuẩn trích dẫn
2.1 ASTM D 1757 Than và cốc - Phương pháp xác định lưu huỳnh trong tro.
3. Tóm tắt phương pháp
3.1 Đốt than hoặc cốc trong điều kiện tiêu chuẩn đến khối lượng không thay đổi
Chuẩn bị hai dung dịch từ tro. Dung dịch A có được bằng cách nung chảy tro với xút (NaOH), rồi hòa tan phần nóng chảy đó trong axit clohidric pha loãng (HCl). Dung dịch B có được bằng cách phân hủy tro trong axit sunfuric (H2SO4), axit fluoric (HF) và axit nitric (HNO3). Dung dịch A dùng để phân tich SiO2 và Al2O3. Dung dịchB dùng để phân tích các thành phần còn lại.
3.2 Hai dung dịch được phân tích bằng sự phối hợp các phương pháp:
1) phương pháp quang phổ dung cho SiO2, Al2O3, TiO2 và P2O5;
2) phương pháp chuẩn độ Chela tometric dung cho CaO và MgO;
3) phương pháp quang kế ngọn lửa dùng cho Na2O và K2O.
Trong hình 1 nêu những nét tổng quát của các phương pháp và cách tiến hành cho từng phép xác định.
4. Ý nghĩa và sử dụng
4.1 Phân tích thành phần của tro thường có ích trong việc mô tả chất lượng chung của than. HIểu biết về thành phần tro cũng có ích khi dự đoán tác động của tro và xỉ trong buồng cháy. Sự sử dụng tro – sản phẩm phụ của quá trình đốt than đôi khi phụ thuộc vào thành phần hóa học của tro.
4.2 Điều chú ý là thành phần hóa học của tro than thí nghiệm có thể không tương ứng với thành phần chất khoáng có trong than, hoặc với thành phần của tro bay và xỉ do đốt than ở qui mô thương mại.
5. Thiết bị
5.1 Cân, chính xác đến 0,1 mg.
5.2 Chén nung – chén niken, dung tích 50 cm3 được dùng để nung chảy tro trong NaOH và chén platin 30 cm3 để phân hủy tro trong HF.
5.3 Thiết bị quang kế ngọn lửa.
5.4 Lò muf đốt nóng bằng điện, có tuần hoàn khí tốt và có thể duy trì nhiệt độ khoảng 750oC.
5.5 Quang phổ kế hấp thụ - vùng hoạt động từ 380 đến 780 nm.
5.6 Rây 150 và 250 µm (số 100 và số 60) tiêu chuẩn Mỹ.
6. Độ tinh khiết của hóa chất và vật liệu
6.1 Độ tinh khiết của hóa chất – chỉ sử dụng các hóa chất thuộc loại thuốc thử trong mọi thử nghiệm. Trừ khi có chỉ định khác, mọi thuốc thử phải phù hợp với quy định trong phân tích. Do điều kiện có thể sử dụng các loại khác nhưng phải được khẳng định đầu tiên là nó có độ tinh khiết khá cao, cho phép sử dụng mà không làm giảm độ chính xác của việc xác định.
6.2 Độ tinh khiết của nước – nếu không có quy định gì khác thì, những điều liên quan tới nước sẽ được hiểu là nước cất hoặc nước có độ tinh khiết tương đương.
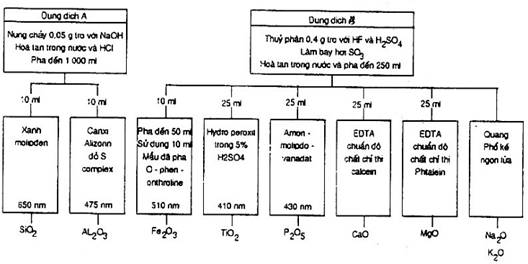
Hình 1 – Nét tổng quát các phương pháp nhanh để phân tích tro của than và cốc
7. Chuẩn bị tro than và tro cốc
7.1 Cách tiến hành – chuẩn bị 3 g đến 5 g tro từ than hoặc cốc được làm khô không khí và trộn đều, được nghiền đến lọt sàng 250 µm (N060). Rải than hoặc cốc thành lớp dầy không quá 6 mm (1.4 insơ) trong lòng đĩa bằng đất sét nung hoặc chịu nhiệt.
Đặt đĩa vào lò muf nguội, tăng dần, sao cho đạt nhiệt độ 5000C trong 1 giờ và lên đến 7500C trong 2 giờ. Đốt cháy đến khối lượng không đổi ( ± 0,001 g, chú thích 2) tại 7500C. Để nguội rồi chuyển vào cối mã não, nghiền đến lọt sàng 150 µm (No100). Nung lại tro ở 7500C trong 1 giờ, làm nguội nhanh, và cân ngay phấn tro đem phân tích. Nếu các mẫu được lưu giữ, thì đốt lại tro trước khi cân hoặc xác định lượng mất khi nung ở 7500C đối với một mẫu riêng biệt được cân cùng lúc với mẫu phân tích. Trộn cẩn thận từng mẫu trước khi cân.
Chú thích 2 – Cốc có thể được đốt đến khối lượng không đổi, tạo nhiệt độ không vượt quá 9500C nếu thấy khó đốt thành tro.
8. Chuẩn bị các dung dịch phân tích (mẫu, mẫu chuẩn, mẫu trắng)
8.1 Thuốc thử
8.1.1 Axit clohidric (1 + 1) pha 1 thể tích axit clohidric đậm đặc (HCl, d 1,19) với 1 thể tích nước.
8.1.2 Axit fluohidric (d 1,15) – Axit flohidric đậm đặc (HF).
8.1.3 Soda fenspat – số 99a. Mẫu của phòng tiêu chuẩn quốc gia
8.1.4 Axit nitric (d 1,42) – Axit nitric đậm đặc (HNO3).
8.1.5 Natri hidroxit, (NaOH) – loại viên nén.
8.1.6 Axit sunfuric (1 + 1), trộn cẩn thận bằng que khuấy, 1 thể tích axit sunfuric đậm đặc (H2SO4 – d 1,84) với 1 thể tích nước.
8.2 Cách tiến hành
8.2.1 Các phương pháp mô tả là cho mẫu tro điển hình, tuy nhiên, sự pha loãng khác nhau hoặc lượng mẫu trích ra khác với qui định này, có thể thường được dùng hơn để có được nồng độ thích hợp đối với cường độ tiêng cho các thành phần khác nhau. Các mầu sắc thay đổi như quy định là ổn định nếu không có những thể hiện gì khác. Tuy rằng các phương pháp mô tả là dùng cho một mẫu đơn, nhưng một nhóm 10 mẫu hoặc nhiều hơn, có thể đucợ xử lý đồng thời. Mỗi giai đoạn được gắn số sẽ được hoàn thành cho một nhóm mẫu đang được phân tích trước khi làm tiếp bước tiếp theo.
8.2.2 Dung dịch A để xác định SiO2 và Al2O3 – Cân 0,0500 g mẫu và chuyển vào chén niken có thể tích 50 cm3. Thêm 1,5 g NaOH, đậy nắp chén, và đốt trong ngọn lửa gas tới khi NaOH nóng chảy. Lắc nhẹ để đảm bảo không có hạt mẫu nào nổi trên bề mặt dung dịch nóng chảy. Tiếp tục nung chảy khoảng 5 phút tại nhiệt độ mẫu đỏ đục, sau đó, đưa chén ra khỏi ngọn lửa và lắc nhẹ dung dịch để nguội dần cho đến khi nó phủ một lớp lên thành chén. Thêm khoảng 25 cm3 nước vào dung dịch, chờ ít nhất 1 giờ hoặc có điều kiện thì để qua đêm. Chuyển dung dịch trong chén vào một cốc dung tích 60 cm3 có chứa 400 cm3 nước và 20 cm3 HCl (1 + 1). Không để chén niken tiếp xúc với axit. Dùng thanh gạt cao su, gạt mọi chất cặn lắng ra khỏi chén và chuyển nó sang cốc. Rửa sạch bình định mức 1 000 cm3 bằng HCl (1 + 1), tráng lại bằng nước, rồi đổ đung dịch trong cốc vào bình đó. Pha nước đến vạch mức 1 000 cm3 và lắc.
8.2.2.1 Dung dịch chuẩn để xác định SiO2 và Al2O3 – chuẩn bị hai dung dịch chuẩn theo cùng một cách (8.2.2), bằng cách dùng các phần 0,0500 g mẫu NIST 99a-Soda fenspat (8.1.3). Đồng thời chuẩn bị dung dịch trắng như 8.2.2, nhưng không dùng Soda fenspat. Chứa các dung dịch chuẩn và dung dịch trắng bằng các chai chất dẻo.
8.2.3 Dung dịch B để xác định FeO, TiO2, P2O5, CaO, MgO, Na2O và K2O. Xử lý 0,400 g ± 0,0005 g mẫu vào chén platin có dung tích 30 cm3 có chứa 3 cm3 H2SO4 (1 + 1) và 10 cm3 HF. Để bay hơi trên bếp cho đén lúc HF bay hơi hết, sau đó thêm 1 cm3 HNO3 và đun nóng tiếp cho đến khi khí SO3 đậm đặc bốc ra. Làm nguội chén và các chất chứa trong đó, thêm nước để hòa tan cặn, và thủy phân trên bếp khoảng 1/2 giờ. Chuyển chất chứa trong chén vào bình định mức 250 cm3. Để nguội tới nhiệt độ phòng, rồi pha loãng tới vạch mức 250 cm3 và lắc đều (chú thích 3). Cũng chuẩn bị một dung dịch trắng B (8.2.3), và không bỏ mẫu vào. Vì có khả năng chất kiềm bị nhiễm bẩn do thủy tinh làm bình, nên sau khi chuẩn bị dung dịch B, cần phải xác định cả natri và kali trong cùng một ngày, hoặc dùng ống pipet hút ra 25cm3 và trữ nó trong chai bằng chất dẻo.
Chú thích 3 – Mặc dù có một lượng sunfat canxi (CaSO4) trong các mẫu chứa nhiều canxi không thể tan được trong quá trình thủy phân trong chén, nhưng nó thường được hòa tan khi pha loãng tiếp. Một lượng nhỏ bari sunfat (BaSO4) không tan có thể còn được giữ lại với một ít mẫu. Nó sẽ lắng xuống đáy bình và không làm cản trở việc xác định thành phần chính. Nếu thấy rõ có cặn lắng không tan thì có thể tiến hành lọc dung dịch vào bình định mức 250 cm3 khác trước khi pha loãng lần cuối để chuẩn độ.
9. Độ chính xác và thiên sai
9.1 Độ lặp lại
Kết quả của các lần xác định liên tiếp tiến hành trong cùng phòng thí nghiệm cùng với một loại thiết bị và do cùng một người thực hiện không được khác nhau vượt quá số phần trăm như sau :
SiO2 1,0 TiO2 1,0 MgO 0,3
Al2O3 0,7 P2O5 0,05 Na2O 0,1
Fe2O3 0,3 CaO 0,2 K2O 0,1
9.2 Độ tái lặp
Trị số trung bình các kết quả của phép xác định hai lần tiến hành ở các phòng thí nghiệm khác nhau trên các phần chia từ cùng một mẫu phân tích, không được khác nhau vượt quá số phần trăm như sau:
SiO2 2,0 TiO2 0,25 MgO 0,5
Al2O3 2,0 P2O5 0,15 Na2O 0,3
Fe2O3 0,7 CaO 0,40 K2O 0,3
9.3 Thiên sai
Không thể xác định được thiên sai.
SILIC DIOXIT (SiO2)
10. Thuốc thử
10.1 Dung dịch amoni môlipdat (7,5 g/100 cm3) – Hòa tan 7,5 g amôni môlipdat [(NH4)6MO7O24.4H2O] vào 75 cm3 nước. Thêm 10 cm3 H2SO4 (1+1) và pha loãng đến 100 cm3. Lưu giữ trong bình chất dẻo.
10.2 Dung dịch khử
Hòa tan 0,7 g natri sunfit (Na2SO3.7H2O) trong 10 cm3 nước. Thêm 0,15 g 1-animo-2-naphthol-4-axit sunforic và khuấy cho tan. Hòa tan 9,0 g dung dịch natri metabisunfit (Na2S2O5) trong 90 cm3 nước. Đổ dung dịch này vào dung dịch trên rồi trộn. Chứa trong chai chất dẻo. Phải kiểm tra tính chất khử của dung dịch dựa vào chất chuẩn đã biết, trừ khi dung dịch mới được chuẩn bị.
10.3 Dung dịch axit tartric (10% trong nước) – chứa trong chai chất dẻo.
11. Cách tiến hành
11.1 Hút 10 cm3 dung dịch trắng và 10 cm3 của mỗi dung dịch chuẩn và dung dịch mẫu A vào các bình định mức riêng 100 cm3. Pha loãng mỗi dung dịch đến 50 cm3 hoặc 60 cm3 với nước rồi lắc đều. Dùng pipet chia độ lấy 1,5 cm3 dung dịch amoni môlipdat cho thêm vào dung dịch trên, lắc đều và để yên trong 10 phút. Dùng ống pipet lấy thêm 4 cm3 dung dịch tartric axit và cho tiếp 1 cm3 dung dịch khử vào bình thử nhất, lắc nhẹ trong lúc cho thêm. Pha loãng dung dịch trong bình này đến vạch mức 100 cm3 và lắc đều trước khi tiến hành các bình tiếp theo. Để mỗi dung dịch yên trong 1 giờ, sau đó xác định độ hấp thụ của nó ở 650 nm bằng cách sử dụng dung dịch trắng làm dung dịch đối chứng, thừa nhận nó có độ hấp thụ bằng 0.
12. Tính toán
12.1 Tính các hệ số f1và f2cho mỗi dung dịch chuẩn như sau:
f1 = CS/A1 và f2 = CS/A2(1)
trong đó
CS là hàm lượng SiO2 trong mẫu chuẩn, tính bằng %;
A1 và A2 là độ hấp thụ đối với các dung dịch chuẩn, mỗi dung dịch chứa 50 mg mẫu chuẩn.
12.2 Tính tỷ lệ phần trăm silic dioxit trong tro như sau:
SiO2, % = FA (2)
Trong đó
F = (f1 + f2)/2;
A là độ hấp thụ của dung dịch mẫu có chứa 50 mg tro.
NHÔM OXIT (Al2O3)
13. Thuốc thử
13.1 Axit axetic, băng (CH3COOH).
13.2 Alizalin đỏ S – dung dịch (0.1% trong nước).
13.3 Dung dịch canxi clorua (CaCl2) (7 g/500 cm3) – chuyển 7g canxi cacbonat (CaCO3) vào cốc 250 cm3. Thêm khoảng 50 cm3 nước và từng giọt HCl (1+1) đến khi CaCO3 được hòa tan. Đun sôi dung dịch từ 1 đến 2 phút, làm nguội rồi pha loãng với nước đến 500 cm3.
13.4 Dung dịch đệm – Hòa tan 70 g natri axetat (CH3COOHNa.3H2O) trong nước. Thêm 30 cm3 CH3COOH rồi pha loãng đến 500 cm3.
13.5 Dung dịch hidroxilamin hidroclorua (10% trong nước) – mới chuẩn bị.
13.6 Axit thioglicolic (5%) – pha loãng 5 cm3 axit thioglicolic với nước đến 100 cm3 mới chuẩn bị.
14. Cách tiến hành
14.1 Lấy 10 cm3 dung dịch mẫu A, 20 cm3 của mẫu dung dịch chuẩn A, 20 cm3 dung dịch trắng vào các bình định mức dung tích 100 cm3 riêng. Sau đó lấy 10 cm3 dung dịch trắng vào bình chứa 10 cm3 dung dịch mẫu để duy trì độ pH cần thiết. Đối với mẫu tro có chứa nhỏ hơn 20% Al2O3 lấy 20 cm3 trực tiếp từ dung dịch mẫu A.
Vừa lắc vừa thêm bằng pipét từng loại thuốc thử theo thứ tự sau: 1 cm3 dung dịch CaCl2, 1 cm3 dung dịch hidroxilamin hidroclorua và 1 cm3 dung dịch axit thioglicolic. Dùng ống đong chia độ thêm 10 cm3 dung dịch đệm cho mỗi bình và để yên trong 10 phút. Dùng pipet thêm 5 cm3 dung dịch alizalin đỏ S, pha loãng đến 100 cm3 rồi lắc. Để mỗi dung dịch yên trong 1 giờ rồi đo độ hấp thụ ở 475 nm sử dụng dung dịch trắng làm mẫu đối chứng, thừa nhận nó có độ hấp thụ bằng 0.
15. Tính toán
15.1 Tính các hệ số f1, f2 cho mỗi phép thử chuẩn như sau:
f1 = CA / A1 và f2 = CA / A2 (3)
trong đó
CA là hàm lượng Al2O3 trong mẫu tenspat chuẩn, tính bằng phần trăm;
A1 và A2 là độ hấp thụ của các dung dịch chuẩn.
15.2 Tỉ lệ phần trăm Al2O3 được tính như sau:
Đối với 10 cm3 dung dịch mẫu A:
Al2O3, % trong tro = 2FA (4)
Đối với 20 cm3 dung dịch mẫu A:
Al2O3, % trong tro = FA (5)
Trong đó
F = (f1 + f2) / 2 và
A là độ hấp thụ của dung dịch mẫu.
SẮT OXIT (Fe2O3)
16. Thuốc thử
16.1 Dung dịch hidroxilamin hidroclorua (10%) – chuẩn bị dung dịch 10% hidroxilamin hidroclorua (NH2OH.HCl) trong nước.
16.2 Ortophenantrolin (0,1%) – chuẩn bị dung dịch 0,1% trong nước.
16.3 Natri xitrat (10%) – chuẩn bị dung dịch 10% Natri xitrat (Na3C8H5O7.2H2O) trong nước.
16.4 Sắt, dung dịch chuẩn (1 cm3 = 0,1 mg Fe2O3) – hoà tan 0,2455 g ± 0,0005 g sắt amôn sunfat [Fe(NH4)2.(SO4)2.6H2O] trong nước, thêm 3 cm3 H2SO4 (1+1) rồi pha loãng đến 500 cm3.
17. Cách tiến hành
17.1 Pha 10 cm3 dung dịch mẫu B đến 50 cm3 trong bình định mức. Lấy 10 cm3 dung dịch mẫu đã pha vào bình định mức 100 cm3, hút 5 cm3 dung dịch sắt chuẩn vào bình định mức 100 cm3 khác và không thêm gì vào bình thứ ba đối với mẫu trắng. Dùng ống đong chia độ, thêm 5 cm3 dung dịch hidro xilamin hidroclorua vào mỗi bình rồi để yên trong 10 phút. Sau đó dùng ống đong chia độ thêm 10 cm3 dung dịch ortophenantrolin vào mỗi bình và khuấy. Cuối cùng dùng ống đong chia độ thêm 10 cm3 dung dịch natri xitrat cho mỗi bình, pha loãng với nước đến 100 cm3 rồi lắc. Sau một giờ, đo độ hấp thụ ở 510 nm, sử dụng mẫu trắng làm mẫu đối chứng và thừa nhận nó có độ hấp thụ bằng 0.
18. Tính toán
18.1 Tính tỉ lệ phần trăm Fe2O3 như sau:
Fe2O3 , % = (A.CF/A1.B) x 100 (6)
trong đó
A là độ hấp thụ của mẫu;
A1 là độ hấp thụ của dung dịch sắt chuẩn;
B lã mẫu chứa trong lần pha loãng cuối cùng của dung dịch B (8.2.3 và 17), tính bằng miligam;
C1 là lượng Fe2O3 trong lượng sắt chuẩn đã lấy, tính bằng miligam.
Chú thích 4 - Đối với các lần pha loãng đã nêu 5 cm3 dung dịch chuẩn sắt chứa 0,5 mg Fe2O3 (CF) và dung dịch mẫu đã pha chứa 3,2 mg mẫu (B). Đối với các mẫu chứa lớn hơn 15% Fe2O3 lấy lượng 5 cm3 dung dịch mẫu đã pha trong đó chứa 1,6 mg mẫu.
TITAN DIOXIT (TiO2)
19. Thuốc thử
19.1 Hidropeoxit (3%) – chuẩn bị dung dịch 3% hidropeoxit (H2O2) trong nước.
19.2 Titan, dung dịch chuẩn (1 cm3 = 0,20 mg TiO2) – chuyển 0,2008 g ± 0,0005 g mẫu NIST số 154a (99,6% TiO2) hoặc số lượng thích hợp TiO2 của thuốc thử có thành phần đã biết vào chén platin. Nung chảy với kali pyrosunfat (theo tỉ lệ khoảng 4 ± 1). Hoà tan dung dịch trong 50 cm3 H2SO4 (1+1) nguội, rồi pha loãng đến 1000 cm3 trong bình định mức.
20. Lập đường chuẩn
20.1 Cho 5,0 cm3 dung dịch chuẩn titan vào bình định mức 50 cm3 số 1 rồi thêm khoảng 25 cm3 nước. Cho 25 cm3 nước vào bình thứ 2. Thêm 5 cm3 H2SO4 (1+1) vào mỗi bình rồi làm nguội đến nhiệt độ phòng. Thêm 5 cm3 H2O2 vào bình số 1, đổ nước vào cả hai bình đến mức 50 cm3 rồi lắc. Đo độ hấp thụ của dung dịch ở bình số 1 ở 410 nm và sử dụng thuốc thử trắng ở bình số 2 làm mẫu đối chứng, thừa nhận nó có độ hấp thụ bằng 0.
21. Cách tiến hành
21.1 Lấy một lượng 25 cm3 dung dịch mẫu B vào bình định mức 50 cm3 (N° 1) và 25 cm3 dung dịch mẫu B vào bình định mức khác (N° 2) để làm mẫu đối chứng. Thêm 5 cm3 H2SO4 (1+1) vào mỗi bình lắc và để nguội đến nhiệt độ phòng. Sau đó thêm 5 cm3 H2O2 vào bình N° 1, thêm nước đến vạch vào cả hai bình và lắc đều. Đo độ hấp thụ của dung dịch trong bình N° 1 ở 410 nm và sử dụng dung dịch trong bình N° 2 làm mẫu đối chứng, thừa nhận độ hấp thụ của nó bằng 0.
22. Tính toán
22.1 Tính tỉ lệ phần trăn TiO2 như sau:
TiO2 , % = (ACT / A1B) x 100 (7)
trong đó
A là độ hấp thụ của dung dịch mẫu;
A1 là độ hấp thụ của dung dịch titan chuẩn;
B là lượng mẫu chứa trong lần pha loãng cuối cùng của dung dịch B, tính bằng miligam;
CT là lượng TiO2 trong lượng dung dịch titan chuẩn đã lấy, tính bằng miligam.
Chú thích 5 - Đối với những lần pha loãng như đã nêu CT = 1 mg TiO2 và B = 40 mg mẫu. Sử dụng các số liệu này, phương trình trên rút gọn thành:
TiO2 , % = 2,5 A/A1
PHỐTPHO PENTOXIT (P2O5)
23. Thuốc thử
23.1 Dung dịch molipdivanadat – hoà tan 0,625 g amôni metavanadat (NH4VO3) trong 200 cm3 HNO3 (1+1) (1 thể tích HNO3 d 1,42 và 1 thể tích nước). Hoà tan 25 g amôni molipdat [(NH4)6Mo7O24.4H2O] trong 200 cm3 nước. Đổ dung dịch amôni môlipdat vào dung dịch metavanadat trong khi đang khuấy, rồi pha nước đến 500 cm3.
23.2 Photpho pentoxit, dung dịch chuẩn đậm đặc – hoà tan với nước 0,3835 g ± 0,0005 g kali dihidro photphat (KH2PO4) đã sấy khô ở 110°C, rồi pha với nước đến 1000 cm3.
23.3 Photpho pentoxit, dung dịch chuẩn làm việc – pha 50 cm3 dung dịch chuẩn đậm đặc đến 500 cm3 với nước, Mỗi 25 cm3 của dung dịch này chứa 0,50 mg P2O5.
24. Cách tiến hành
24.1 Lấy 25 cm3 dung dịch mẫu B, 25 cm3 dung dịch chuẩn làm việc và 25 cm3 dung dịch trắng vào các bình định mức 50 cm3 riêng. Dùng pipet thêm 10 cm3 dung dịch molipdivanadat vào mỗi bình, pha với nước đến vạch mức và để yên trong 5 phút. Đo độ hấp thụ ở 430 nm sử dụng mẫu trắng làm mẫu đối chứng, thừa nhận nó có độ hấp thụ bằng 0.
25. Tính toán
25.1 Tính tỷ lệ phần trăm P2O5 như sau:
P2O5 , % = (ACP / A1B) x 100 (8)
trong đó
A là độ hấp thụ của dung dịch mẫu;
A1 là độ hấp thụ của dung dịch photpho chuẩn;
B là lượng mẫu chứa trong lần pha loãng cuối cùng của dung dịch B, tính bằng miligam;
CP là lượng P2O5 trong lượng dung dịch photpho chuẩn đã lấy, tính bằng miligam.
Chú thích 6 – Đối với những lần pha loãng như đã nếu: CP = 0,5 mg và B = 40 mg mẫu. Sử dụng các số liệu này phương trình nêu trên rút gọn thành:
P2O5 , % = 1,25 A/A1
CANXI OXIT (CaO) và MANHE OXIT (MgO)
26. Thuốc thử
26.1 Amonihidroxit (d 0,90) – amonihidroxit đậm đặc (NH4OH).
26.2 Chỉ thị calcein – Trộn đều 0,2 calcein, 0,12 g thimolphtalein và 20 g kaliclorua (KCl) đã nghiền mịn.
26.3 Dung dịch EDTA – hoà tan 3,720 g muối dinatri của axit etylen diamine tetraaxetic (EDTA) trong nước và pha đến 1000 cm3. Chuẩn lại với dung dịch canxi chuẩn sử dụng chỉ thị calcein hoặc phtalein đỏ.
26.4 Axit clohidric (d 1,19) – axit clohidric đậm đặc (HCl).
26.5 Axit clohidric (1+1) – trộn 1 thể tích HCl đậm đặc (d 1,19) với 1 thể tích nước.
26.6 Chỉ thị phtalein đỏ - trộn 0,1 g phtalein đỏ, 0,005g metyl đỏ và 0,05 g xanh naphton B với 10g KCl đã nghiền mịn.
26.7 Dung dịch kali hidroxit (224,4 g/l) – hoà tan 224,4 g kali hidroxit (KOH, 85%) trong nước và pha đến 1000 cm3. Giữ trong chai chất dẻo.
26.8 Dung dịch canxi chuẩn (1000 g/l) (1 cm3 = 0,00056 g CaO) – hòa tan 1,000 g canxi cacbonat (CaCO3) trong nước với 4 cm3 HCl (1+1). Đun nóng đến sôi để đuổi cacbon dioxit (CO2), làm nguội rồi pha đến 1000 cm3.
26.9 Dung dịch triethanolamin – pha 500 cm3 triethanolamin với nước đến 1000 cm3 và khuấy.
27. Cách tiến hành với CaO
27.1 Lấy 25 cm3 dung dịch mẫu B và 25 cm3 dung dịch trắng vào bình chuẩn độ 500 cm3 riêng rẽ. Pha với nước đến khoảng 100 cm3. Thêm theo trình tự 20 giọt HCl đậm đặc, 5 cm3 dung dịch triethanolamin, 5 cm3 NH4OH vaf 10 cm3 dung dịch KOH và lắc đều sau khi thêm mỗi thuốc thử. Pha với nước đến khoảng 200 cm3. Thêm khoảng 40 mg chỉ thị calcein và chuẩn độ với dung dịch chuẩn EDTA đến khi mầu thay đổi từ huỳnh quang xanh sang đỏ tía. Quan sát sự thay đổi màu trong ánh sáng khuyếch tán xuống qua bình đến bề mặt đen.
28. Cách tiến hành với MgO
28.1 Lấy 25 cm3 dung dịch mẫu B và 25 cm3 dung dịch trắng vào bình chuẩn độ riêng rẽ. Pha với nước đến khoảng 100 cm3. Thêm 20 giọt HCl đậm đặc, 20 cm3 dung dịch triethanolamin và 25 cm3 NH4OH, khuấy sau khi thêm mỗi thuốc thử. Pha với nước đến khoảng 200 cm3. Thêm một thể tích dung dịch chuẩn EDTA hơi ít hơn khi chuẩn canxi. Sau đó thêm khoảng 40 mg chỉ thị phtalein đỏ. Tiếp tục chuẩn độ tới khi màu thay đổi từ đỏ nhạt đến không mầu hoặc xám nhạt. Một lượng dư nhỏ dung dịch EDTA gây ra mầu xanh lá cây. Quan sát sự đổi mầu trong ánh sáng khuyếch tán xuống qua bình tới bề mặt trắng.
29. Tính toán
29.1 Tính tỷ lệ phần trăm CaO như sau:
CaO, % = x 100 (9)
trong đó
V1 là thể tích dung dịch EDTA cần để chuẩn độ canxi trừ đi thể tích dung dịch EDTA cần để chuẩn độ mầu trắng, tính bằng centimét khối;
F là lượng CaO tương đương với 1 cm3 EDTA, tính bằng gam;
W là lượng mẫu, tính bằng gam;
A là thể tích dung dịch mẫu đã lấy, tính bằng centimét khối.
Chú thích – Nếu W = 0,4 g, F = 0,0056 g và A = 25 cm3 thì tỷ lệ phần trăm của CaO = 1,4V1.
29.2 Tính tỷ lệ phần trăm MgO như sau:
MgO, % = x100 (10)
trong đó
V2 là thể tích của dung dịch EDTA cần để chuẩn độ canxi cộng với manhê trừ đi thể tích dung dịch EDTA cần để chuẩn độ mẫu trắng, tính bằng centimét khối;
0,719F là lượng MgO tương đương với 1 cm3 EDTA, tính bằng gam;
Các ký hiệu còn lại giống như khi tính toán tỷ lệ phần trăm CaO.
NATRI OXIT (Na2O) VÀ KALI OXIT (K2O)
30. Thuốc thử
30.1 Kali, dung dịch đậm đặc (100 phần triệu) – hoà tan 0,2228 g ± 0,0005 g kali sunfat (K2SO4) trong nước và pha đến 1000 cm3.
30.2 Kali, dung dịch chuẩn (50 phần triệu) – lấy 50 cm3 dung dịch kali đậm đặc vào bình định mức 100 cm3. Thêm 10 cm3 dung dịch tro tổng hợp và 10 cm3 dung dịch dữ trữ natri, pha nước đến vạch mức và khuấy. Dung dịch chứa 50 phần triệu kali. Dung dịch natri chuẩn 10 phần triệu chứa 10 phần triệu kali, nên cũng sử dụng nó như dung dịch kali chuẩn 10 phần triệu. Giữ trong chai chất dẻo.
30.3 Natri, dung dịch đậm đặc (100 phần triệu) – hoà tan trong nước 0,3088 g ± 0,0005 g natri sunfat khô (Na2SO4) và pha đến 1000 cm3.
30.4 Natri, dung dịch chuẩn (10 và 50 phần triệu) – lấy 10 cm3 và 50 cm3 dung dịch đậm đặc natri vào bình định mức 100 cm3 riêng biệt. Thêm 10 cm3 dung dịch tro tổng hợp và 10 cm3 dung dịch đậm đặc kali vào mỗi bình, pha nước đến vạch mức 100 cm3 và lắc đều. Những dung dịch này chứa 10 và 50 phần triệu.
30.5 Dung dịch tro tổng hợp
30.5.1 Hoà tan 2,0 sắt trong 10 cm3 trong axit sunfuric (H2SO4) (1+1) và 100 cm3 nước.
30.5.2 Hoà tan 10 g nhôm trong 10 cm3 axit sunfuric (H2SO4) (1+1) và 100 cm3 nước.
30.5.3 Hoà tan 1,25 g CaCO3 trong 3 cm3 axit sunfuric (H2SO4) (1+1) và 600 cm3 nước.
30.5.4 Hoà tan 1,0 g manhe sunfat (MgSO4.7H2O) trong nước.
30.5.5 Gộp chung các dung dịch từ 30.5.1 đến 30.5.4, lọc và pha với nước đến 1000 cm3.
31. Lập đường chuẩn
31.1 Chuẩn bị các dung dịch chuẩn natri chứa 1, 4, 20, 30, và 40 phần triệu natri và các dung dịch chuẩn kali chứa 1, 4, 20, 30, và 40 phần triệu kali. Chúng được chuẩn bị bằng cách lấy những lượng thích hợp từ các dung dịch đậm đặc (30.1 và 30.3) và tiến hành như đã mô tả ở điều 30.2 và 30.4. Chuẩn bị các dung dịch trắng, dung dịch trắng natri chứa 10 cm3 dung dịch tro tổng hợp và 10 cm3 dung dịch dữ trữ kali trên 100 cm3, dung dịch trắng kali chứa 10 cm3 dung dịch tro tổng hợp và 10 cm3 dung dịch chứa natri trên 100 cm3. Rửa từng chén mẫu hai lần với dung dịch mà chén sẽ chứa trước khi rót dung dịch thử. Sau khi điều chỉnh ảnh quang phổ ngọn lửa theo chỉ dẫn của nhà chế tạo, đẻ bước sóng là 589 nm và hút dung dịch natri 50 phần triệu. Di chuyển bước sóng kiểm tra xung quanh bước sóng natri để tìm độ nhạy cực đại. Cuối cùng, trong khi hút dung dịch natri 50 phần triệu, điều chỉnh khe hở và độ nhậy để đạt được chỉ số truyền ánh sáng 80%. Rửa máy hút bằng nước rồi xác định chỉ số truyền của dung dịch natri 40 phần triệu. Lặp lại động tác này để kiểm tra độ truyền sáng là 80% của dung dịch natri 50 phần triệu và tiến hành với các dung dịch natri 30, 20 và 10 phần triệu. Thử với dung dịch trắng và trừ đi chỉ số của mẫu trắng từ mỗi phép xác định. Dựng đường cong làm việc của dung dịch trong khoảng từ 10 đến 50 phần triệu. Để làm việc này, hút dung dịch natri 10 phần triệu và tăng chiều rộng khe hở đủ để đọc được điểm giao chuyển 80, sau đó xác định các điểm đối với dung dịch natri 4 và 1 phần triệu. Làm các phép đo tương tự đối với dung dịch chuẩn kali ở bước sóng 768 nm và vẽ đồ thị các số liệu thu được.
32. Cách tiến hành
32.1 Vận hành máy quang kế ngọn lửa để điều chỉnh theo cách dùng. Rửa chén mẫu bằng dung dịch mà chén sẽ chứa, rót dung dịch chuẩn natri 50 và 10 phần triệu, dung dịch mẫu B, dung dịch trắng và nước vào các chén riêng biệt. Điều chỉnh dụng cụ ở bước sóng dung dịch natri khoảng 10 đến 50 phần triệu (589 nm). Sử dụng dung dịch chuẩn natri 50 phần triệu, xác định các điểm đọc đối với dung dịch mẫu và dung dịch trắng. Khi dung dịch mẫu chứa natri ít hơn 10 phần triệu phải đọc ở khoảng từ 1 đến 10 phần triệu sau khi đã điều chỉnh thiết bị theo dung dịch natri 10 phần triệu. Đối với mẫu giàu natri, pha một lượng dung dịch B để thử. Thực hiện quy trình tương tự ở 768 nm đối với kali sử dụng dung dịch chuẩn kali 50 và 10 phần triệu để điều chỉnh quang phổ kế. Khấu trừ điểm trắng từ mỗi mẫu và xác định nồng độ natri và kali (CN và CK) trong dung dịch thử từ đường cong làm việc thích hợp.
32.2 Khi sử dụng kính lọc của quang kế ngọn lửa để xác định natri, chú ý là quang phổ của nó tạo nhiễu. Nồng đồ 5 + 1 của kali đến natri và nồng độ 10 + 1 của canxi đến natri gây ra nhiễu quang phổ có thể kết quả hàm lượng natri sẽ tăng rõ rệt ở tuyến natri 589 nm.
33. Tính toán
33.1 Tính tỷ số phần trăm của Na2O như sau:
Na2O , % = CN x 1,348/16 (11)
trong đó
CN là nồng độ natri khi sử dụng 0,4 g mẫu trong 250 cm3 dung dịch B, tính bằng phần triệu;
33.2 Tính tỉ lệ phần trăm của K2O như sau:
K2O , % = CK x 1,2046 / 16 (12)
trong đó
CK là nồng độ kali khi sử dụng 0,4 g mẫu trong 250 cm3 dung dịch B, tính bằng phần triệu.
Chú thích 8 - Nếu CN = 10 phần triệu, natri chứa trong 250 cm3 = 0,0025 g có nguồn gốc từ 0,4 g tro, vậy:
[(phần triệu Na x Na2O/2Na)/ phần triệu mẫu] x 100 = Na2O, %
Hoặc Na2O, % = [(0,0025)(1,348)/0,4 x 100 = 0,84
16 yếu tố sử dụng trong các phương trình trên đã nhận được từ các loại tính toán và cho phép sử dụng “phần triệu” trực tiếp không qua chuyển đổi thành “gam” natri và kali.
34. Những từ chính
34.1 Nhôm oxit (Al2O3); canxi oxit (CaO); thành phần nguyên tố tro; sắt oxit (Fe2O3); quang kế ngọn lửa; manhe oxit (MgO); photpho pentoxit (P2O5); kali oxit (K2O); silic dioxit (SiO2), natri oxit (Na2O); titan dioxit (TiO2)
1) Mẫu soda feldspat chứa 65,2% SiO2 và 20,5% Al2O3 chỉ dùng cho phân tích.